Rare Diseases 101 – Fabry Disease

Our commitment to improving the diagnostic odyssey for those affected by rare diseases goes beyond our diagnostic applications, with our disease education blog series we hope to raise awareness and understanding.
In this post, the first of our series, we talk about Fabry disease. With new treatment options on the horizon there is a glimmer of hope for those affected by Fabry.
What is Fabry Disease?
Fabry Disease is a rare Inherited Metabolic Disorder belonging to a group known as ‘Lysosomal Storage Disorders’.
Understanding Fabry disease
What is an Inherited Metabolic Disorder?
Our bodies are continuously performing a metabolic process – a collection of chemical reactions that take place in our cells converting our food into energy. This energy is responsible for everything we do, from physical movement to brain function and growth. Cells use Enzymes in order to speed up the process of producing, recycling and removing vital compounds fast enough to sustain life.
What are Lysosomal Storage Disorders?
Lysosomes are the ‘recycling centres’ of cells. They contain enzymes that are responsible for digesting (breaking down) certain compounds. Mutations in the genes that encode these enzymes are responsible for the family of genetic diseases called lysosomal storage disorders.
Fabry disease is caused by mutations (or alterations) in the a-Gal A gene (GLA) that instructs cells to make the a-Gal A enzyme. The a-Gal A enzyme is responsible for breaking down specific complex sugar-lipid molecules, specifically, globotriaosylceramide (GL-3 or Gb3), lyso-GL-3/Gb3 and related glycolipids.
The enzyme deficiency causes a continuous build-up of the fatty GL-3/Gb3 and related glycolipids in the body’s cells, mainly affecting the blood and the walls of the blood vessels throughout the body. Over time, this builds up causes the blood vessels to become narrow particularly affecting the skin, liver, heart, brain and the nervous system.
What are the differences between the two types of Fabry Disease?
There are two marked types (or subdivisions), Type 1 classic phenotype which manifest during childhood and type 2 later-onset phenotype, both having serious consequences such as renal failure, cardiac disease and even early death. In the classic manifestation of the disease (type 1) symptoms include ‘Angiokeratoma’ (dark red, spotted skin rash), reduced ability to sweat, pain in hands and feet and clouding of the corneas. Patients with later-onset may experience, gastrointestinal problems (nausea, vomiting) and cardiovascular problems (hear failure, arrhythmia).
How Fabry disease affects you? - Signs and Symptoms
Most common symptoms
- Acroparesthesias - episodes of severe pain, particularly in the hands and feet
- Angiokeratomas - small reddish/blue lesions on the skin
- Hypohidrosis or Anhidrosis - decreased or no ability to sweat
- Clouding of cornea/corneal dystrophy - twisting of the blood vessels at the back of the eye (does not affect vision)
- Hearing problems/Tinnitus
- Gastrointestinal problems - cramping and increased frequency of bowl movements particularly after eating
Other less common symptoms
Due to progressive accumulation of the glycolipid in the vascular system, renal, cardiac and cerebrovascular complications may not be seen until later in life although it can be evident in childhood. These complications can lead to heart and kidney failure and stroke.
Fabry disease mnemonic

What is the inheritance pattern and prevalence of Fabry Disease?
Fabry disease occurs universally across all ethnic groups and at an estimated incidence of one in 40,000 of the male population and one in 20,000 of the female population.
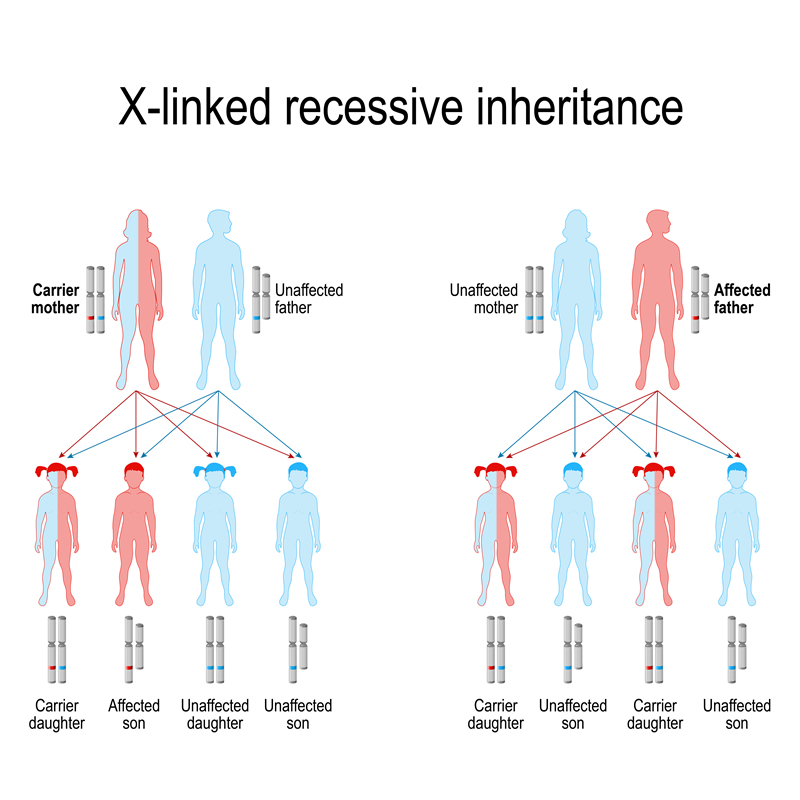
The inheritance pattern of Fabry Disease is called X-Linked Inheritance. The defective GLA gene is on the X-chromosome, one of two chromosomes that determine sex. Females have two X chromosomes, one inherited from their mother and one from their father. Males have one X chromosome inherited from their mother and one Y chromosome inherited from their father.
Girls and boys of an affected mother both have a 50% chance of inheriting the defective gene. Fathers carrying the Fabry gene will pass on the defective gene to girls only.
Affected individuals by type and gender
|
Fabrys Disease Type |
Males |
Females |
|---|---|---|
|
Type 1 |
1/40000 |
1/20000 |
|
Type 2 |
1/1500-4000 |
1/750-2000 |
How is Fabry Disease Diagnosed?
With a wide spectrum of symptoms that vary from person to person, and the rare incidence of Fabry disease, diagnosis is difficult and often delayed and mis-diagnosed. This is particularly the case in children where the pain associated with Fabry disease is often mistaken for growing pains. Recognising the early stage symptoms of pain, skin lesions and reduced or absent sweating can be key to early diagnosis.
How is Fabry Disease Tested?
Tests confirming the enzyme deficiency in males or identifying the GLA Gene mutation in both males and females are used to confirm a Fabry diagnosis.
Prenatal diagnosis of Fabry disease is possible by measuring the a-Gal A activity in amniotic fluid surrounding the foetus and may be offered to expectant mothers who have Fabry disease. Specialist genetic advice and counselling should be sought with regards to any prenatal testing.
What Treatments are available for Fabry Disease?
With no cure, those with Fabry disease may be clinically managed symptomatically to prevent the effects of Fabry’s disease for example, to manage pain associated with this disease patients may be prescribed with oral drugs such as ‘Carbamazepine’ or ‘Diphenylhydantoin’.
In addition, Enzyme Replacement Therapy may be prescribed.
Enzyme Replacement Therapy (ERT)
Enzyme Replacement Therapy (ERT) was approved for Fabry Disease in 2003 and uses a genetically engineered form of the a-Gal A enzyme manufactured from cultures of cells which have been altered to express the human enzyme. This enzyme is administered via intravenous infusion to the patient, usually every two weeks.
Studies have been promising, with improvement for individuals undergoing ERT showing reduced pain, stabilised kidney function and stabilised or improve cardiac abnormalities, much improving quality of life. It is hoped that early treatment will help to prevent the adverse health effects of the disease.
While these have shown to have positive effects on kidney and heart manifestations at an early phase of the disease, long term clinical benefits are unclear. Cost of Fabrazyme is at $200,000(or £179796 approx. converted).
Treatments and research in development
Oral chaperone therapy
Chaperone or Enzyme Enhancement Therapy aims to find medicines that stabilise abnormal a-galactosidase enzymes, made by some people with Fabry disease, to allow the enzyme to function enough to prevent lysosomal storage in cells.
A study using a pharmacological chaperone has completed its phase two trial– to determine drug safety and is undergoing phase three – to determine effectiveness. This is being carried out at several international sites including two within the UK. If successful a new oral tablet treatment may be available as a treatment option to people with this particular mutation.
For more information on clinical trials visit www.clinicaltrials.gov
Fabry Disease related organisations
There are a number of charitable organisations that can offer support to those affected by Fabry Disease. For more information please visit:
The MPS Society (UK)
http://www.mpssociety.org.uk/diseases/fabry/
Metabolic Support UK (UK)
https://www.metabolicsupportuk.org/
Fabry Support and Information Group (USA)
http://www.fabry.org/FSIG.nsf/Pages/About
References
Nord https://rarediseases.org/rare-diseases/fabry-disease/
The MPS Society http://www.mpssociety.org.uk/diseases/fabry/
Metabolic Support UK https://www.metabolicsupportuk.org/wp-content/uploads/2017/09/Fabry-Disease.pdf
Fabry Support and Information Group http://www.fabry.org/FSIG.nsf/Pages/Fabry
If you liked this article maybe you will also find interesting the following in-depth articles about other rare diseases, like Speaker Series - Niemann-Pick Disease Type C Speaker Series - Adrenoleukodystrophy (ALD) Disease Mendelian & Modality NHS Partnership